 官方网站-首页官方网站-首页
官方网站-首页官方网站-首页
新闻中心

发布时间:2025-11-07 16:00:27
阅读量:275次
2025年诺贝尔物理学奖颁给“电路中宏观量子隧穿效应和能量量子化”的三位发现者。这一发现证明量子效应可以存在于宏观系统,突破了我们对经典世界的固有认知。
听起来不可思议的隧穿效应,其实并不仅仅存在于物理学家的精密电路。在化学世界里,它同样扮演着“特立独行”的角色——分子体系的能量即使不足以翻越势垒(lěi),也(yě)可(kě)能(néng)“瞬(shùn)移(yí)”般(bān)穿(chuān)透(tòu)它(tā),从(cóng)而(ér)极(jí)大(dà)地(de)加(jiā)速(sù)反(fǎn)应(yīng)进(jìn)程(chéng)。量子隧穿距离我们并不遥远,它甚至参与了人体内的酒精代谢——无论是临窗小酌,抑或与朋友开怀畅饮,量子隧穿都可能在我们的肝脏里悄然发生。
撰文 | 郑超(中国科学院上海有机化学研究所研究员)
老马识途:哈蒙德假说
波兰尼和艾林的过渡态理论识别出势能面上的关键点,揭示了分子体系结构、能量与反应速率常数之间的内在关联。然而“从头推导”的化学理论在实际应用时再次遇到了“维度爆炸”的难题。在原子共线的氢交换反应中,只有两个独立变化的核坐标,当然可以仿照真实的地形等高线图,在一张白纸上复刻出反应的势能面;同时考虑到体系的对称性,该反应过渡态的原子排布也是容易猜想的。但是,复杂一些的情况又该如何处理呢?一般地,由N个原子构成的分子体系总共有3N个自由度。扣除质心的三个平动自由度、以及分子整体绕其主轴旋转的三个转(zhuǎn)动(dòng)自(zì)由(yóu)度(dù)之(zhī)后(hòu),还(hái)剩(shèng)3N–6个(gè)自(zì)由(yóu)度(dù)对(duì)应(yīng)分(fēn)子(zi)的(de)内(nèi)振(zhèn)动(dòng)(线(xiàn)性(xìng)分(fēn)子(zi)体(tǐ)系(xì)只(zhǐ)有(yǒu)两(liǎng)个(gè)转(zhuǎn)动(dòng)自(zì)由(yóu)度(dù),故(gù)内(nèi)振(zhèn)动(dòng)的(de)自(zì)由(yóu)度(dù)的(de)数(shù)目为3N–5)。换句话说,化学反应势能面的维度高达3N–6(或者3N–5)。面对如此复杂的结构空间,想要依靠纯数学的方法建立完整的反应势能面,并计算出过渡态的能垒和速率常数,无疑是极为困难的。那么,能否在此过程中融入适当的化学见解,从而对(至少一部分反应的)过渡态的能量和结构进行合理的估计呢?
G. S. Hammond (1921~2005)
1955年1月1日,《美国化学会志》刊登了一篇题为“反应速率的相关性”(A correlation of reaction rates)的长文,文章唯一的作者是美国爱荷华州立大学的助理教授哈蒙德(G. S. Hammond)。在这篇文章中,哈蒙德提出了一个后来用他的姓氏命名的假说,从有机化学的角度提供了一把理解分子体系结构和能量关系的钥匙。哈蒙德假说指出:对于反应路径上的两个相邻状态(比如过渡态和与之连接的原料或产物),如果它们的能量非常接近,那么它们的结构差异也必然十分微小。这条论断用朴素的化学语言勾勒出势能面理应满足的一项数学性质——连续性。事实上,我们有理由提出更强的要求:势能面在任意自由度上必须二阶可偏导,且导函数连续。同时,反应路径的曲率及其变化率不能过大,以避免出现陡峭的“悬崖”或者“断层”。考虑到过渡态的能量总是高于原料和产物,那么从哈蒙德假说的原始表述可以立即推出以下结论:对于放热反应(原料的能量高于产物),过渡态的能量和结构应当更接近原料,因此可以形象地称作“早期”过渡态。与之相反,在吸热反应(产物的能量高于原料)中,过渡态的能量和结构应当与产物更接近,是为“晚期”过渡态。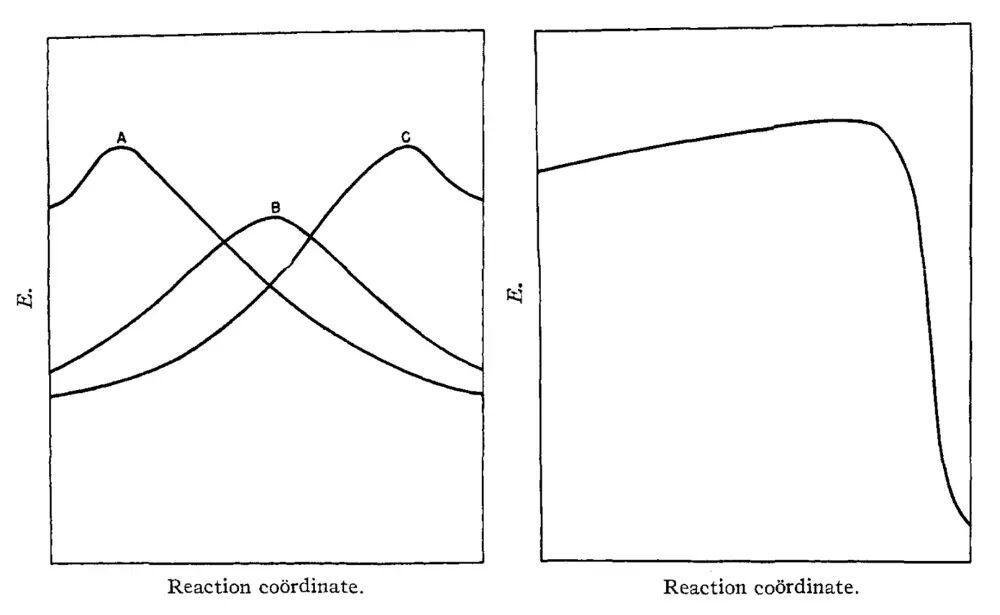
图7.哈蒙德原始论文中关于能量和反应坐标的示意图。(左)A为放热反应中结构和能量与原料接近(jìn)的(de)早(zǎo)期(qī)过(guò)渡(dù)态(tài),C为(wèi)吸(xī)热(rè)反(fǎn)应(yīng)中(zhōng)结(jié)构(gòu)和(hé)能(néng)量(liàng)与(yǔ)产(chǎn)物(wù)接(jiē)近(jìn)的(de)晚(wǎn)期(qī)过(guò)渡(dù)态(tài),B为(wèi)热(rè)中(zhōng)性(xìng)反(fǎn)应(yīng)的(de)过(guò)渡(dù)态(tài),其(qí)能(néng)量(liàng)和(hé)结(jié)构(gòu)不(bù)能(néng)用(yòng)类似的方法推测。(右)反应路径中不应出现的悬崖形貌(剧烈放热反应的过渡态结构与产物接近,但能量与原料接近)。图片来源:J. Am. Chem. Soc.1955, 77, 334.
对于半个多世纪前的有机化学家而言,哈蒙德假说最大的价值是允许他们基于反应的宏观热力学特性(即放热或吸热程度),对无法利用实验观察的过渡态结构进行猜想。而仅仅依靠这些猜想,已经足以得出许多关于重要基元反应机制的真知灼见。这方面最直观的例(lì)
通(tōng)常(cháng)伴(bàn)随着体系能量的升高,是溶剂解反应的决速步。根据哈蒙德假说,其过渡态结构应当与相应的碳正离子接近。当时的有机化学家已经非常熟悉,碳正离子的稳定性随叔碳/仲碳/伯碳/甲基的顺序依次减弱,同时相应的溶剂解反应的速率也随之减慢(伯碳和甲基卤代物甚至无法通过SN1途径发生溶剂解反应)。对于叔碳和仲碳离子的形成过程来说,显然后者的能量升幅更大。如果把哈蒙德假说对过渡态结构和能量关系的预言再向前推进一步,这种能量升幅的差别无疑意味着形成仲碳离子的过渡态更加晚期。如果以断键的碳卤原子间距作为反应坐标,那么可以认为在形成仲碳离子的过渡态中,碳卤原子的间距要比叔碳离子体系更远。这便是基于哈蒙德假说,对卤代烃溶剂解反应的关键过渡态结构做出的一种估计。至于这种估计是否可靠,我们不妨联想一下劈柴的场景。假如木质特别松脆,斧子只需楔进去一点,柴禾就会自己裂开(放热反应/早期过渡态);而当木质十分致密时,得用力劈到底端才能成功(吸热反应/晚期过渡态)。显然,哈蒙德假说蕴含的思想与人们的日常生活经验是一致的。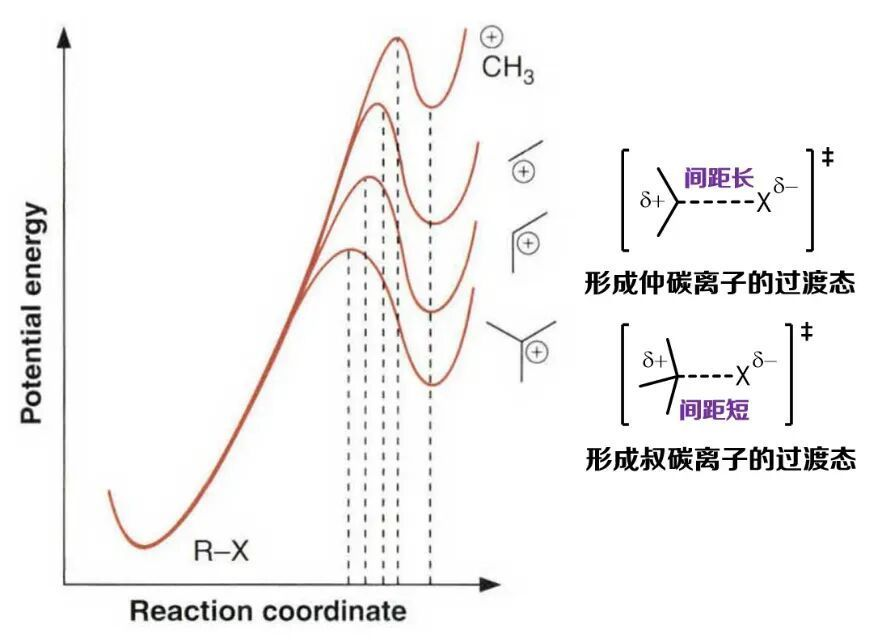
图8. 用哈蒙德假说估计碳正离子形成过程的过渡态能量和结构。左图来源:参考文献[17]。
敏锐的读者朋友一定已经注意到,拓展哈蒙德假说内涵的过程触及了反应热力学性质与动力学行为的内在联系:一般地,如果同类反应的吸热(或放热)程度越高,那么其速率将越慢(或越快)。早在1924年,丹麦化学家布朗斯特(J. N. Brønsted)在研究酸催化酯或醚的水解反应时,首先指出其速率常数k的自然对数与酸催化剂的解离平衡常数pKa的负值线性相关
其中α和β是经验参数。(17)式通常用贝尔、埃文斯和波兰尼三人的姓氏命名,简称BEP方程。(16)和(17)式同属前文介绍过的“线性自由能关系”,揭示了同类反应的热力学驱动力和动力学能垒之间的简明数学关联。这里被再三强调的“同类”的含义(yì)可(kě)以(yǐ)从(cóng)势(shì)能(néng)面(miàn)和过渡态的角度加以理解:即这些反应共享相同的反应坐标和类似的过渡态结构(正如卤代烃溶剂解过程中形成叔碳离子和仲碳离子的(de)两(liǎng)个(gè)反(fǎn)应(yīng))。因此,哈蒙德假说完全可以作为解读BEP方程的直观模型。事实上,(17)式中的参数α(其取值范围一般在0和1之间)恰好起到在反应路径上指示过渡态位置的作用。当α接近0时,活化能几乎不受反应焓变的影响,过渡态结构必然与原料相似;而当α接近1时,活化能与反应焓变几乎同步改变,当然对应于晚期过渡态!
图9. 贝尔(左)和埃文斯、波兰尼(右)关于BEP方程(用蓝线标记)的原始论文。图片来源:Proc. R. Soc. London, Ser. A1936, 154, 414. Trans. Faraday Soc.1936, 32, 1333.
物理有机化学家们从哈蒙德(dé)假(jiǎ)说(shuō)的(de)原(yuán)始(shǐ)表(biǎo)述(shù)中(zhōng)阐(chǎn)发(fā)的(de)思(sī)想(xiǎng)也(yě)许(xǔ)远(yuǎn)远(yuǎn)超(chāo)过(guò)了(le)他(tā)的(de)本(běn)意(yì)。哈(hā)蒙(méng)德(dé)学(xué)术(shù)生(shēng)涯(yá)中(zhōng)更(gèng)多(duō)的(de)贡(gòng)献(xiàn)集中(zhōng)于(yú)有(yǒu)机(jī)光(guāng)化(huà)学(xué),他(tā)是(shì)二十世纪中期将物理有机化学的理念引入光化学领域的代表性人物之一。1959年哈蒙德与超分子化学家克拉姆(D. J. Cram,1987年(nián)诺(nuò)贝(bèi)尔(ěr)化(huà)学(xué)奖(jiǎng)获(huò)得(de)者(zhě))合(hé)著(zhe)了(le)一(yī)部(bù)在(zài)美(měi)国(guó)产(chǎn)生(shēng)广(guǎng)泛(fàn)影(yǐng)响(xiǎng)的(de)《有(yǒu)机(jī)化学》教科书。有趣的是,在这本书中找不到关于哈蒙德假说的任何讨论,但是哈蒙德假说在有机化学理论体系中的地位是无法撼动的。2003年,为纪念《美国化学会志》创刊125周年,美国化学会旗下的《化学与工程新闻》杂志评选出了曾在《美国化学会志》上发表的125篇最具影响力的论文。哈蒙德假说的原始论文高居第(dì)15位(wèi)。
随着分子电子结构理论的成熟和计算机硬件性能的提升,计算有机化学日益成为有机化学家研究化学动力学的强大工具(用哈特里–福克的流程求解给定核坐标下分子体系的能量,已经能在主流配置的个人电脑上用Gaussian等程序轻松实现了)。确定目标反应的过渡态结构,即搜寻反应路径上的一阶鞍(ān)点(diǎn),是(shì)用(yòng)计(jì)算(suàn)有(yǒu)机(jī)化(huà)学(xué)研(yán)究(jiū)反(fǎn)应(yīng)动(dòng)力(lì)学(xué)和(hé)选(xuǎn)择(zé)性(xìng)问(wèn)题(tí)的(de)核(hé)心(xīn)步(bù)骤(zhòu)。在(zài)Gaussian程(chéng)序(xù)中(zhōng),这(zhè)通(tōng)常(cháng)依(yī)靠(kào)内(nèi)置(zhì)的(de)伯(bó)尼(ní)算(suàn)法(fǎ)(Berny algorithm)来(lái)实(shí)现(xiàn)。该(gāi)算(suàn)法(fǎ)通(tōng)过(guò)递(dì)推(tuī)修(xiū)正(zhèng)势(shì)能(néng)面(miàn)的(de)二(èr)阶(jiē)偏(piān)导(dǎo)数(shù)矩(ju)阵(zhèn)(海(hǎi)森(sēn)矩(ju)阵(zhèn),Hessian matrix)
猜结构必须落在预期过渡态附近的二次区域内(使得初始海森矩阵有且仅有一个负本征值)。换句话说,即使在计算化学的范式下,化学家对过渡态结构的正确估计仍然是解决动力学问题的一个先决条件。从这个意义上讲,哈蒙德假说并未过时,它像一匹识途的老马,始终不倦地指引我们向过渡态进发。
特立独行:化学反应中的量子隧穿
亲爱的读者朋友,读到这里,你是否已经确信过渡态是所有化学反应的必经之路,并且其能垒总是与反应的焓变正相关?很遗憾,这并不正确。在势能面上还有不少“特立独行”的现象,让化学动力学的故事格外丰富多彩。
当我们用“曲面上翻滚的小球”来类比化学反应中分子体系结构和能量的变化,并且默认这枚运动的小球总是在曲面上留下连续的轨迹时,相当于在头脑中构建了一幅由经典力学支配的物理图像。然而,微观世界中有不同寻常的量子效应。根据海森堡不确定性原理(Uncertainty principle),微观粒子不可能同时具备确切的位置x和动量p,且其不确定度满足ΔxΔp ≥ h/4π。以(yǐ)绕(rào)原(yuán)子(zi)核运动的电子为例,若能确定其在某时刻的坐标,则必然无法确定其在该时刻的速度。那么下一时刻这个电子会出现在何处,便无从知晓了。因此从薛定谔方程解出的电子波函数ψ(r)并不能对应一条体现电子运动轨迹的连续曲线。事实上,根据玻恩对波函数的统计诠释,波函数的模方|ψ(r)|^2正比于电子在r处出现的概率密度,而中学化学课本中的“电子云”图案恰是此概率密度分布的一种形象写照。
R. P. Bell(1907~1996)
分子体系的尺度远远大于电子,以分子为主角的化学反应中是否会出现类似的量子效应呢?答案是肯定的,分子在势能面上的轨迹完全可以不连续,这就是化学反应中的量子隧
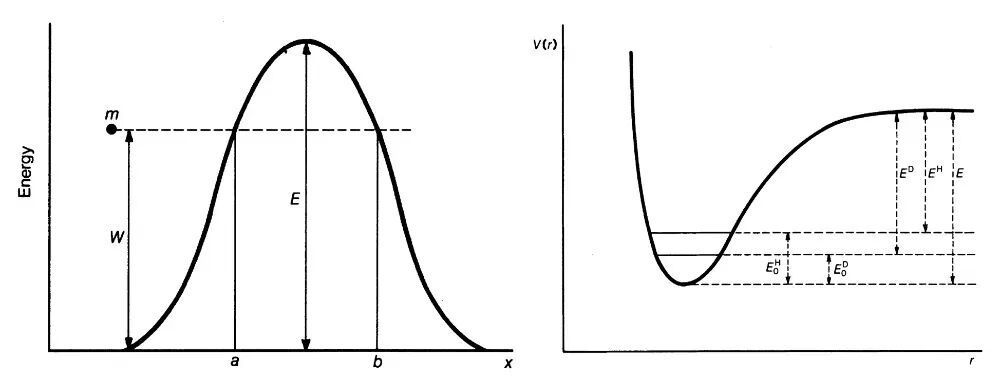
图10.(左)能量为W的粒子m穿透高度为E的势垒的示意图,势垒宽度为b–a。(右)碳–氢(氘)键的解离曲线示意图,曲线底部的两条水平线段表示碳–氢(氘)键伸缩振动的零点能。图片来源:参考文献[16]。
量子隧穿是从第一性原理导出的严格结论。无论它多么不合“常理”,只要我们认同量子力学的基本假设,就必须接纳它的存在。问题在于,量子隧穿能否在化学反应中导致可观测的实验结果呢?1931年,美国化学家尤里(H. C. Urey)分离出氢的同位素氘(D)(他因此获得了1934年诺贝尔化学奖),翻开了同位素化学的新篇章。化学家们很快发现,氢/氘动力学同位素效应是实验观测量子隧穿的绝佳场景。动力学同位素效应源于分子体系的另一种量子特征——零点振动能。在量子力学中如果使用谐振子模型处理分子的内振动,会发现分子即使在绝对零度下也并非完全静止(否则谐振子将同时具备确切的位置和速度,违反不确定性原理),而必然在一定程度上发生“零点振动”,其能量为

测的抛物线型势垒宽仅63.5 pm),可以断定量子隧穿是该氢迁移反应的主要途径。在生命体内的酶促反应中也有量子隧穿的踪迹。1997年,《美国国家科学院院刊》登载了一篇关于马肝乙醇脱氢酶(LADH)催化苄醇氧化反应的论文。研究人员通过测量氘/氚取代的苄醇参与反应的动力学行为,确认了量子隧穿现象的存在;并且发现对LADH活性位点附近的氨基酸残基进行定点突变,还能调控量子隧穿的强弱。考虑到哺乳动物乙醇脱氢酶的结构保守性,不妨想象:无论是独自临窗小酌,抑或与朋友开怀畅饮,量子隧穿都在我们的肝脏中参与酒精的代谢呢!
测的抛物线型势垒宽仅63.5 pm),可以断定量子隧穿是该氢迁移反应的主要途径。在生命体内的酶促反应中也有量子隧穿的踪迹。1997年,《美国国家科学院院刊》登载了一篇关于马肝乙醇脱氢酶(LADH)催化苄醇氧化反应的论文。研究人员(yuán)通(tōng)过(guò)测(cè)量(liàng)氘(dāo)/氚(chuān)取(qǔ)代(dài)的(de)苄(biàn)醇(chún)参(cān)与(yǔ)反(fǎn)应(yīng)的(de)动(dòng)力(lì)学(xué)行(xíng)为(wèi),确(què)认(rèn)了(le)量(liàng)子(zi)隧(suì)穿(chuān)现(xiàn)象(xiàng)的(de)存在;并且发现对LADH活性位点附近的氨基酸残基进行定点突变,还能调控量子隧穿的强弱。考虑到哺乳动物乙醇脱氢酶的结构保守性,不妨想象:无论是独自临窗小酌,抑或与朋友开怀畅饮,量子隧穿都在我们的肝脏中参与酒精的代谢呢!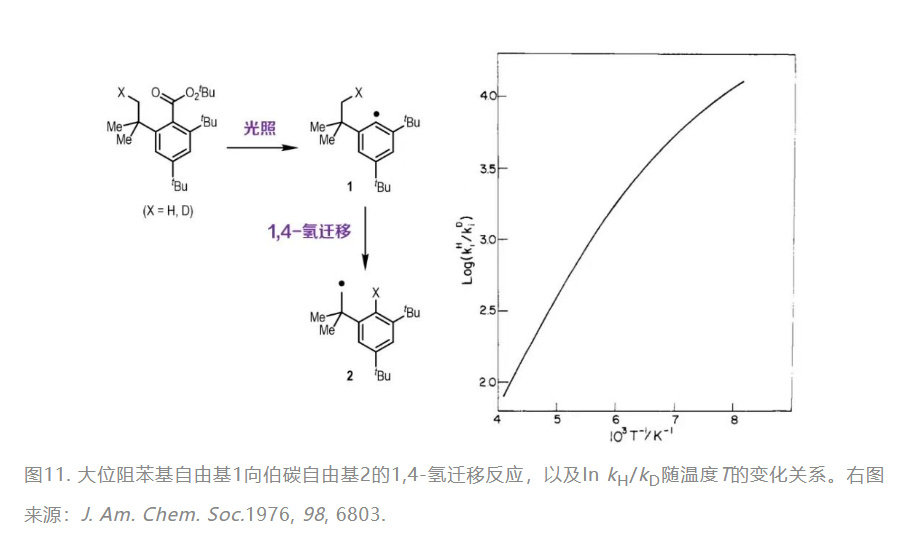
过犹不及:马库斯理论与反转区
R. A. Marcus(1923~)1992年诺贝尔化学奖获得者
行文至(zhì)此(cǐ),我(wǒ)们(men)所(suǒ)遇(yù)到(dào)的(de)化(huà)学(xué)反(fǎn)应(yīng)都(dōu)在(zài)单(dān)一(yī)的(de)势(shì)能(néng)面(miàn)上(shàng)进(jìn)行(xíng);并(bìng)且(qiě)反(fǎn)应(yīng)中(zhōng)伴(bàn)随(suí)着(zhe)化(huà)学(xué)键的(de)断(duàn)裂(liè)和(hé)形(xíng)成,换句话说,我们总能在势能面上标识出一定的反应坐标。但是,还有一大类重要的化学转化——电子转移反应,并不具备上述特征。电子转移在日常生活中无处不在:生命体的呼吸过程、绿色植物的光合作用、金属的锈蚀与防腐、电池的充放电循环等等,电子转移都在其中扮演着重要的角色。对于经历“外球机制”的电子转移反应
其始态(i)和末态(f)的电子结构迥异(电中性或自由基离子),处在不同的势能面上,因此无法直接套用过渡态理论计算反应的速率常数。溶液中电子转移反应的动力学是二十世纪五十年代物理化学的研究热点。为了解答这个问题,化学家们曾经提出了许多方案。但笑到最后的,是美籍加拿大裔化学家马库斯(R. A. Marcus)的电子转移理论。
马库斯理论的出发点仍然是B–O近似。由于电子运动的速度远快于原子核,电子从供体分子(D)向受体分子(A)转移的“瞬间”,虽然体系的电荷分布已经发生了改变,但是D和A的分子结构、以及周围溶剂分子的排布方式都还来不及做出响应。同时考虑能量守恒的要求,我们可以粗略地将电子转移理解为一种“等构型、等能量”的过程。马库斯把D/A分子体系及其周围溶剂环境的结构变化抽象为一维位型坐标q,并将电子转移反应始末态的能量涨落用两条力常数相等的抛物线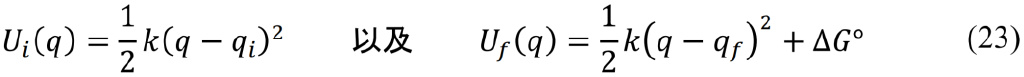
来表示。其中qi和qf分别代表反应始末态的平衡结构,ΔG°是电子转移过程的吉布斯自由能变化(即反应的热力学驱动力,自发过程满足ΔG° < 0)。那么根据等构型、等能量的要求,电子转移一定发生在两条抛物线的交点处,其速率常数可以用量子力学中描述态–态跃迁的费米黄金规则


由于引入了“违背常理”的反转区,化学家们一度对马库斯理论的有效性颇感怀疑。但是,如果将反转区看作马库斯理论的预言,并且从实验上观测到电子转移过程中,热力学驱动力和反应速率之间在一定条件下存在负相关现象,将意味着化学动力学理论和实践的重大突破!在马库斯理论被提出近三十年后的1984年,这项突破终于来临了。美国阿贡国家实验室的米勒(J. R. Miller)和芝加哥大学的克罗斯(G. L. Closs)等人通过一组极富巧思的控制变量实验证实了马库斯理论中反转区的存在。他们将精心挑选的D/A结构单元分别连接在刚性的甾体分子骨架的C16位和C3位。使得目标电子转移反应发生时,D/A结构单元的空间距离被限制在1000 pm左右,并且反应的ΔG°值有较大的可变范围(–0.05至–2.40
够进行大幅度地竖向移动,以覆盖潜在的反转区。利用阿贡国家实验室的20 MeV线性加速器所产生的高能电子脉冲引发电子转移反应,同时用吸收光谱测量反应的表观动力学,明确无误地观察到随着ΔG°值(zhí)的持续降低,电子转移反应速率先加快后减慢,而且变化方式与(25)式和(26)式的预测十分吻合。米勒和克罗斯的实验打消了人们对马库斯电子转移理论的疑虑,也成为此后物理有机化学教科书频繁引用的经典案例。1992年,马库斯由于对“化学系统中电子转(zhuǎn)移(yí)反(fǎn)应(yīng)理(lǐ)论(lùn)”的(de)贡(gòng)献(xiàn)获(huò)得(de)了(le)诺(nuò)贝(bèi)尔(ěr)化(huà)学(xué)奖(jiǎng)。
图12. 马库斯的诺贝尔奖演讲中关于电子转移反应速率常数和重组能的公式。图片来源:
https://www.nobelprize.org/。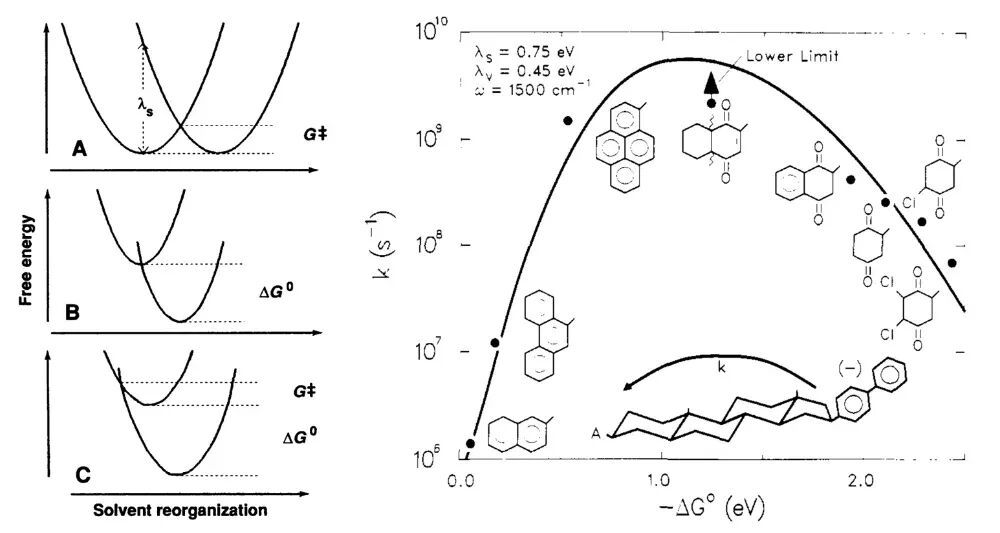

马库斯1923年出生于加拿大蒙特利尔的一个犹太人家庭,1958年归化为美国公民。马库斯提出电子转移理论时,只是一名在布鲁克林理工学院(现已并入纽约大学)获得教职不久的年轻人。后来他又在伊利诺伊大学工作多年,并于1978年加入加州理工学院。获得诺贝尔奖时,马库斯已经年近70岁。他在一场电化学会议中接到来自斯德哥尔摩的电话。在随后临时召开的新闻发布会上,马库斯对诺贝尔奖带来的新声誉感到有(yǒu)些(xiē)困惑。他对记者说:“我不知道我是否希望人们对我的工作有更多关注,我只希望有更多时间来完成它。”他当然有足够的时间!现在(2025年11月)马库斯已满102周岁,但是还保持着相当的学术活跃度:根据谷歌学术搜索的结果,马库斯在最近五年发(fā)表(biǎo)了(le)十(shí)余(yú)篇(piān)论(lùn)文。他是目前仍然在世的最年长的诺贝尔奖获得者。
未完待续……
参考文献
[1] H. B. Schlegel, J. Comput. Chem.1982, 3, 214.
[2] K. J. Laidler, M. C. King, J. Phys. Chem.1983, 87, 2657.
[3] K. J. Laidler, J. Chem. Educ.1985, 62, 1012.
[4] Y. T. Lee, Science1987, 236, 793.
[5] S. Pedersen, J. L. Herek, A. H. Zewail, Science1994, 266, 1359.
[6] B. J. Bahnson, T. D. Colby, J. K. Chin, B. M. Goldstein, J. P. Klinman, Proc. Natl. Acad. Sci. USA1997, 94, 12797.
[7] G. A. Petersson, Theor. Chem. Acc.2000, 103, 190.
[8] J. Van Houten, J. Chem. Educ.2002, 79, 926
[9] C. C. Wamser, J. Phys. Chem. A2003, 107, 18, 3149.
[10] M. J. Nye, J. Comput. Chem.2007, 28, 98.
[11] K.A. Dambrowitz, S.M. Kuznicki. Bull. Hist. Chem.2010, 35, 46.
[12] E. M. Simmons, J. F. Hartwig, Angew. Chem. Int. Ed.2012, 51, 3066.
[13] M. Polanyi, Atomic Reactions. Williams and Norgate, London, 1932.
[14] S. Glasstone, K. J. Laidler, H. Eyring, The Theory of Rate Processes. McGraw-Hill Book Company, 1941.
[15] D. R. Herschbach, Reactive Scattering in Molecular Beams. In Advance in Chemical Physics, Volume X (J. Ross Ed.), John Wiley & Sons. Inc., 1966.
[16] R. P. Bell, The Tunnel Effect in Chemistry. University Press, Cambridge, 1980.
[17] E. V. Anslyn, D. A. Dougherty, Modern Physical Organic Chemistry. University Science Books, 2006.
[18] E. G. Lewars, Computational Chemistry, Introduction to the Theory and Applications of Molecular and Quantum Mechanics. 2nd Ed. Springer, 2011.
[19] R. Monk, Robert Oppenheimer – A Life Inside the Center. Doubleday, 2012.
[20] M. J. Nye, Michael Polanyi and His Generation: Origins of the Social Construction of Science. The University of Chicago Press, 2011.
[21]邓从豪,《中国科学》,1980年,第6期,179页。
[22] 曹则贤,《物理》,2015年,第44卷第3期,193页。
[23] 曹则贤,《物理》,2023年,第53卷第4期,259页。
[24] 钱学森编,《物理力学讲义》(新世纪版),上海交通大学出版社,2007年。
[25] A. A. 福洛斯特、R. G. 皮尔逊著,孙承谔等译,《化学动力学和历程:均相化学反应的研究》,科学出版社,1966年。
[26] M. 玻恩著,李宝恒译,《我的一生和我的观点》,商务印书馆,1979年。
[27] 凯•伯德、马丁• J •舍温著,汪冰译,《奥本海默传》,中信出版集团,2023年。
[28] G. 赫兹堡著,王鼎昌译,《分子光谱与分子结构》(第一卷),科学出版社,1983年。
[29] 艾哈迈德•泽维尔著,陈养正、伍义生、吴应荣、李军译,孔繁敖、胡升华审,《穿越时间的航行——我的诺贝尔奖之路》,科学出版社,2003年。
[30] M. 玻恩、黄昆著,葛惟锟、贾惟义译,江丕桓校,《晶格动力学理论》,北京大学出版社,2011年。
[31] 迈克尔•波兰尼著,徐陶、许泽民译,陈维政校,《个人知识:朝向后批判哲学》(重译本),上海人民出版社,2021年。
[32] 辛德勇,中国历史地理论丛,1989年04期,42页。
[33] 政协陕西省潼关县委员会文史资料委员会编,《潼关文史资料》(第七辑),1994年。
致谢
作者感谢中国科学院上海有机化学研究所游书力院士、中国科学院物理研究所曹则贤研究员、中国科学院大连化学物理研究所傅碧娜研究员、清华大学杨杰教授、美国范德比尔特大学杨中悦教授对本文的宝贵意见。
作者简介
郑超博士,中国科学院上海有机化学研究所研究员,国家自然科学基金委员会优秀青年科学基金项(xiàng)目(mù)获(huò)得(de)者(zhě)。研(yán)究(jiū)方(fāng)向(xiàng)为(wèi)物(wù)理(lǐ)有(yǒu)机(jī)化(huà)学(xué)与(yǔ)手(shǒu)性(xìng)合(hé)成(chéng)。
注(zhù):本(běn)文封(fēng)面(miàn)图(tú)片(piàn)来(lái)自(zì)版(bǎn)权(quán)图(tú)库(kù),转(zhuǎn)载(zài)使用可能引发版权纠纷。
特 别 提 示
1. 进入『返朴』微信公众号底部菜单“精品专栏“,可查阅不同主题系列科普文章。
2. 『返朴』提供按月检索文章功能。关注公众号,回复四位数组成的年份+月份,如“1903”,可获取2019年3月的文章索引,以此类推。
版权说明:欢迎个人转发,任何形式的媒体或机构未经授权,不得(de)转(zhuǎn)载(zài)和(hé)摘(zhāi)编(biān)。转(zhuǎn)载(zài)授(shòu)权(quán)请(qǐng)在(zài)「返(fǎn)朴(pǔ)」微(wēi)信(xìn)公(gōng)众(zhòng)号(hào)内(nèi)联(lián)系(xì)后(hòu)台(tái)。



